TL;DR
CryoSTAR is a cryo-EM analysis tool for resolving continuous conformational heterogeneity directly from particle images.
CryoSTAR has been validated on diverse experimental datasets, including large complexes, membrane proteins, and small proteins.
What can CryoSTAR do for you?
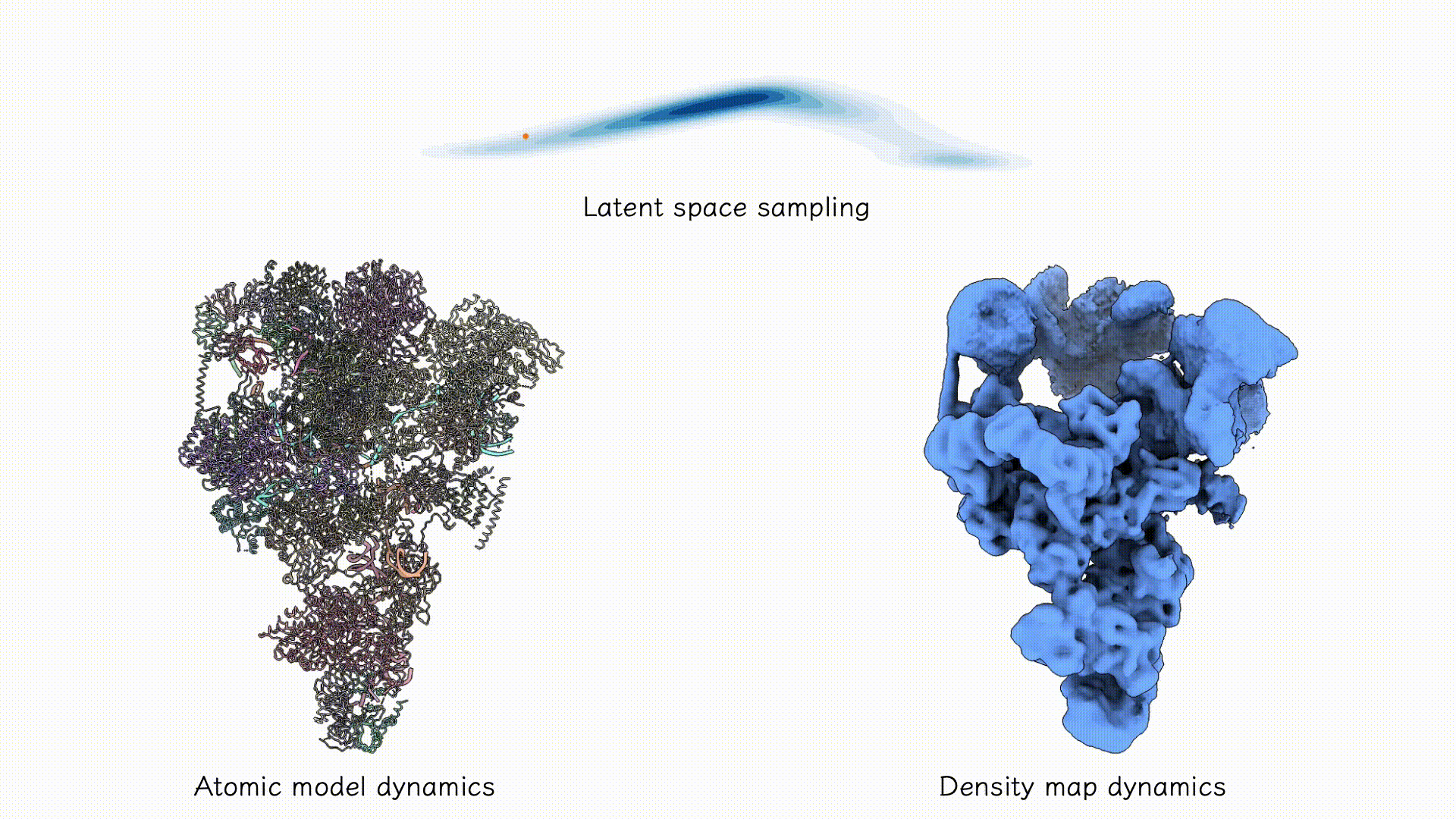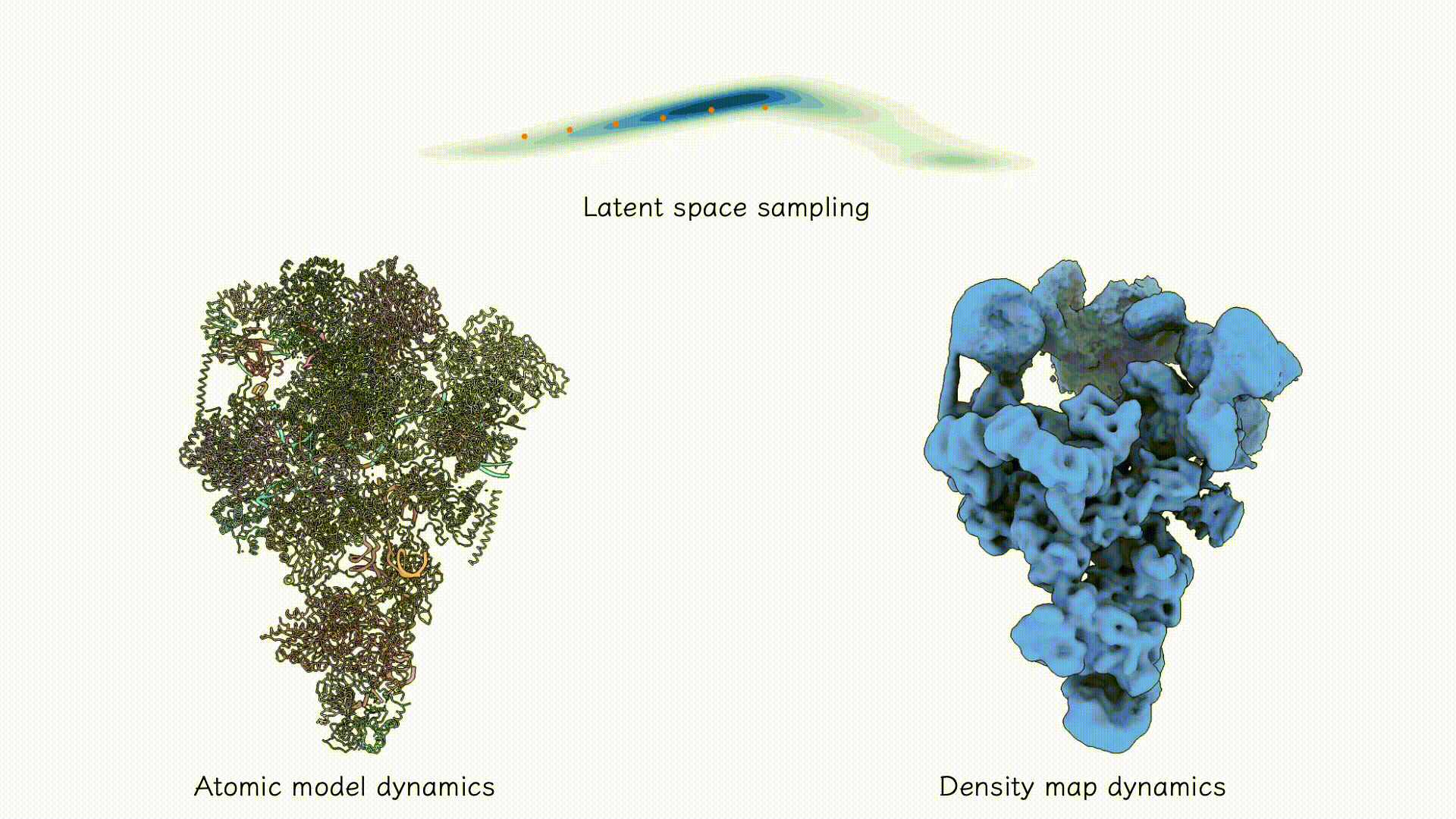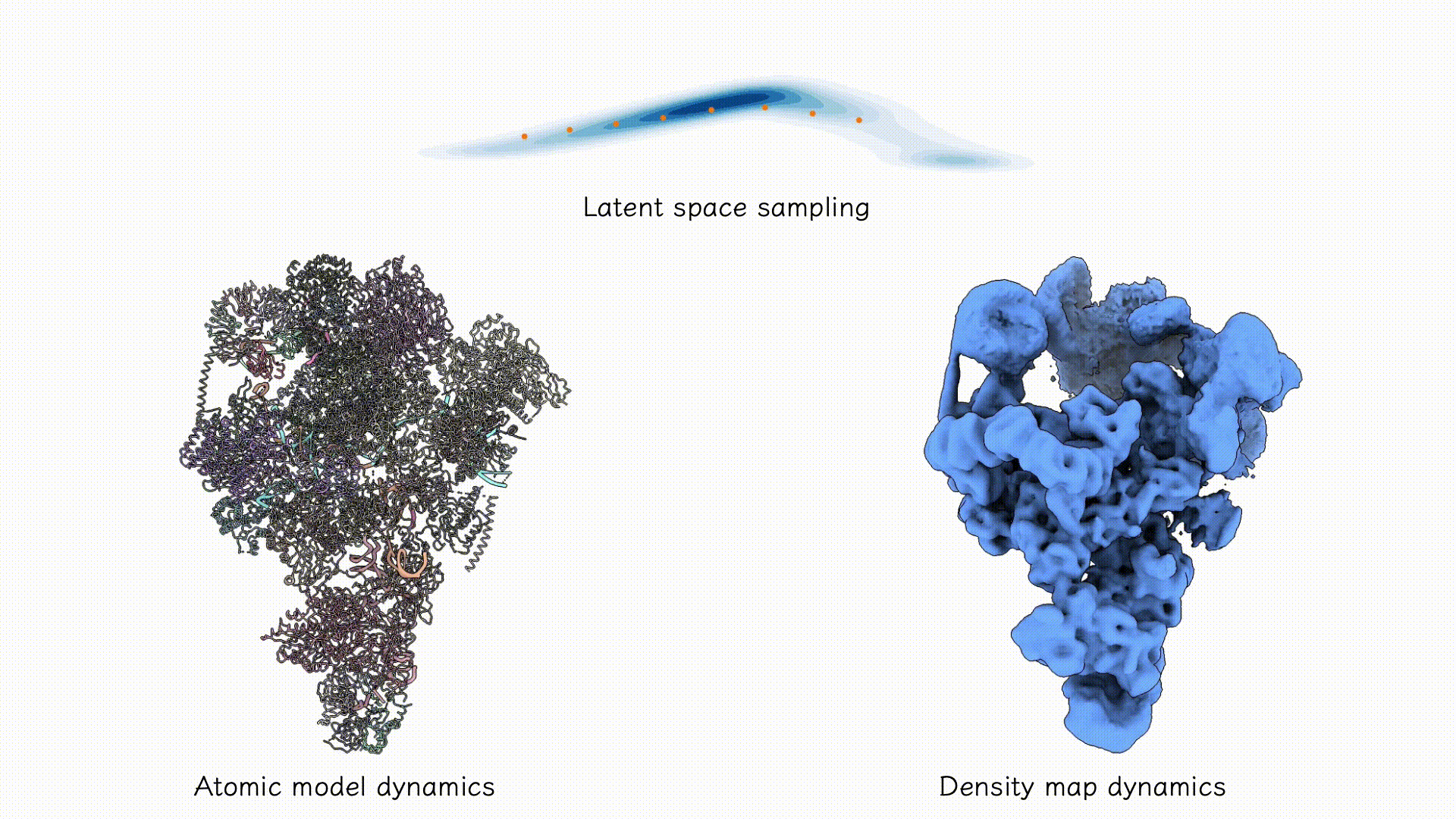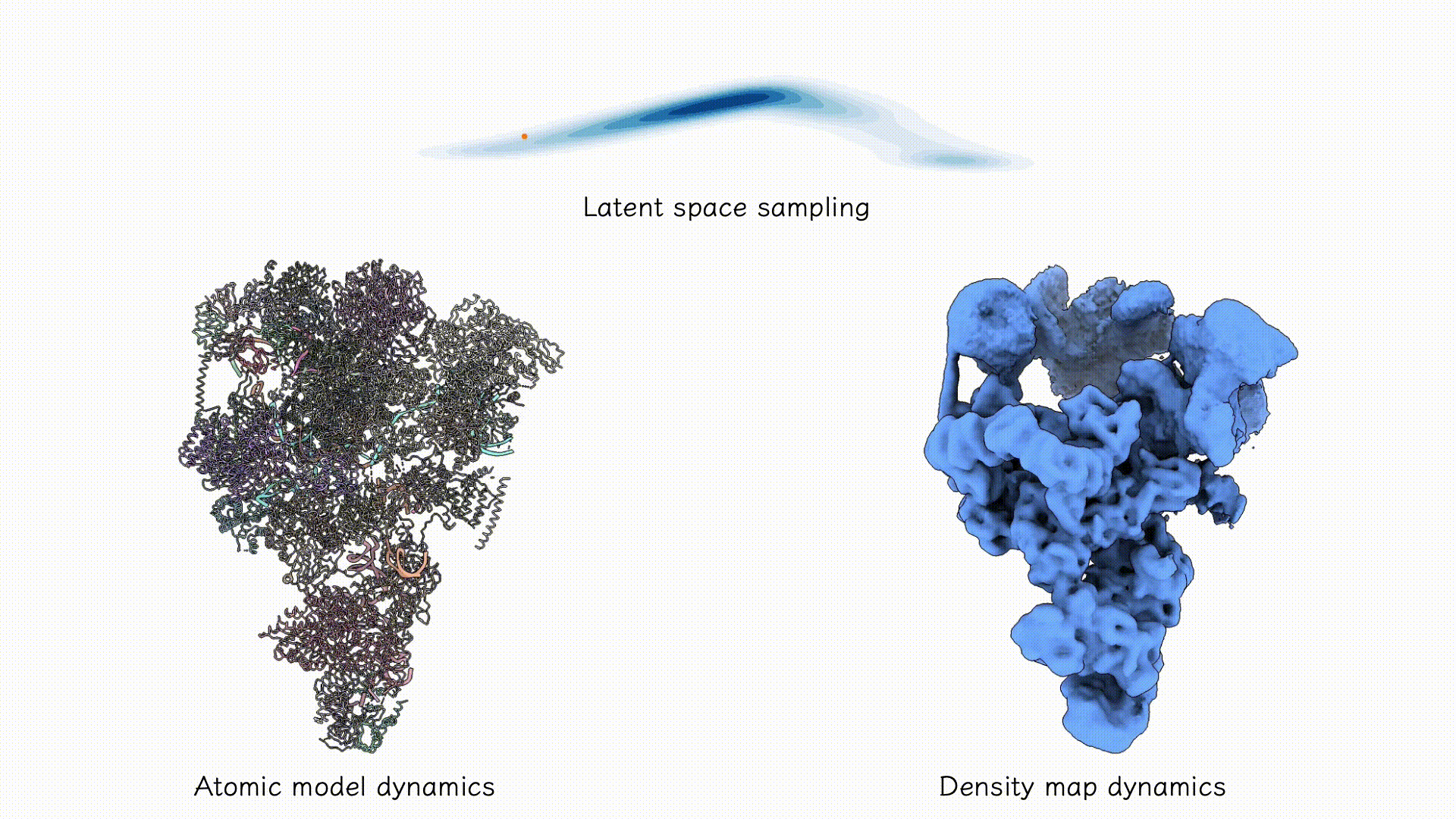
Example on EMPIAR-10180.
With CryoSTAR, you can:
- Reveal continuous motions that are hidden by standard 3D classification
- Obtain both density maps and corresponding coarse-grained structural models
- Assess structural hypotheses using density-based validation (FSC)
CryoSTAR produces two complementary outputs for each conformation:
- Density maps: Generated directly from particle images, these maps remain minimally biased and serve as a reliable reference for interpretation.
- Coarse-grained structural models: Derived from an input atomic model, these models highlight conformational changes in a physically interpretable manner.
Independent density validation is a design choice of CryoSTAR:
Importantly, density maps and structural models are generated through separate stages,
allowing density maps to be used to validate — rather than assume — the inferred motions.
What is cryoSTAR?
The origin of the name “CryoSTAR“ stems from the concept of structural regularization. Our method is a structural constraint-based approach serving the analysis of continuous conformational heterogeneity in cryoEM.
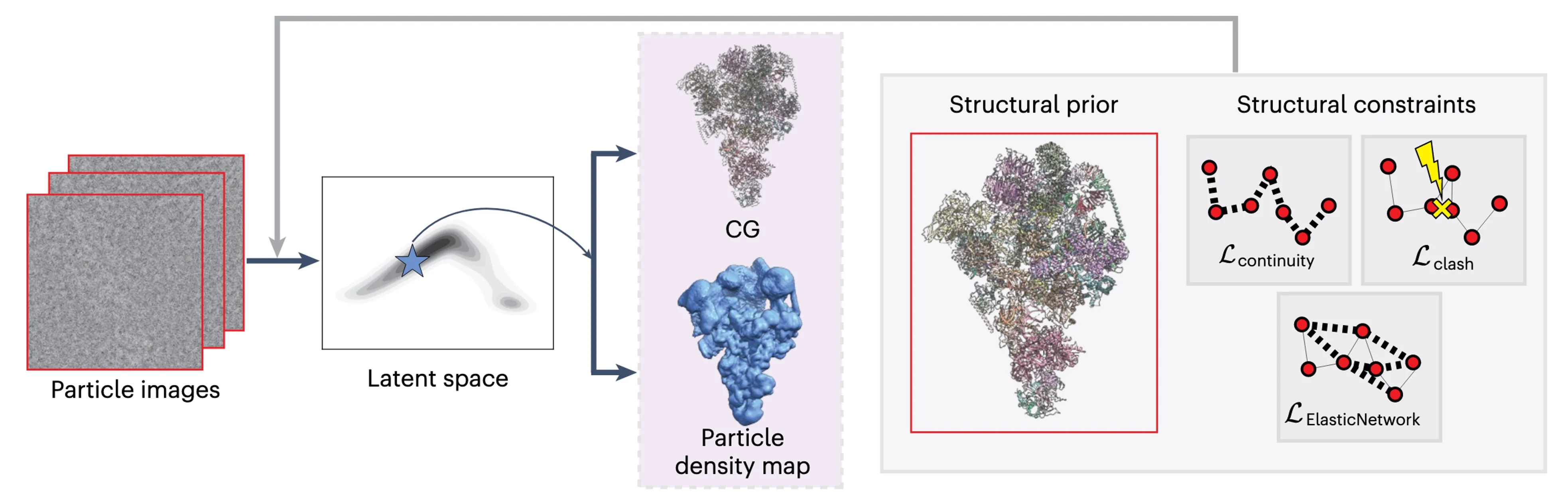
An overview of CryoSTAR.
CryoSTAR takes two complementary inputs to analyze conformational heterogeneity in cryo-EM data:
- A cryo-EM particle dataset of a single macromolecular complex, with particle poses and CTFs estimated by standard upstream methods.
- A single static atomic model of the target, which may come from a homogeneous reconstruction, protein structure prediction, or a known homolog from the PDB.
What CryoSTAR does?
CryoSTAR maps each particle image into a low-dimensional latent space that captures structural variability. From the distribution of latent variables, we infer the population and organization of distinct conformations present in the dataset. Crucially, CryoSTAR also enables image-wise posterior inference: for each particle, we can sample its corresponding conformation from the posterior distribution.
The output includes:
- Coarse-grained atomic models representing different conformations
- Corresponding density maps for structural interpretation
Appropriate structure priors matter
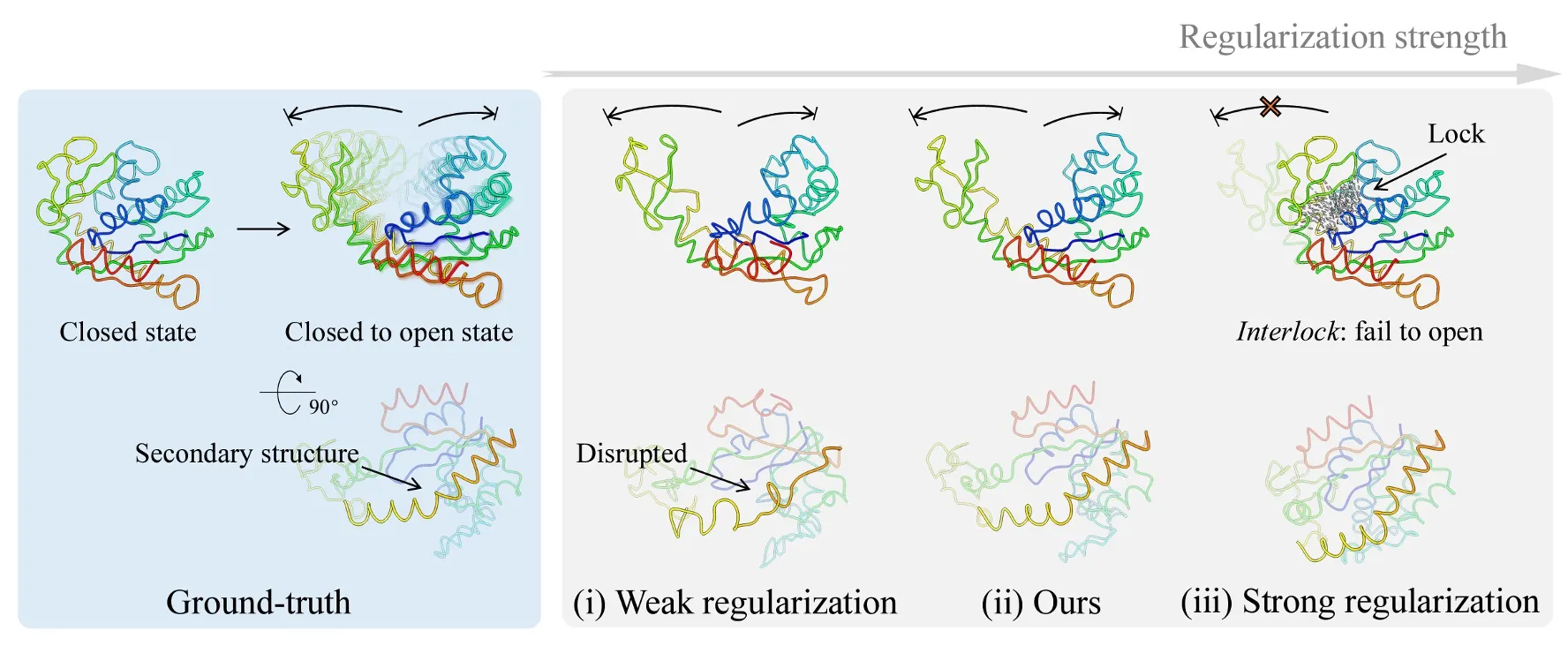
Proper regularization strength is the key to leveraging prior structural information.
CryoSTAR is built around structure-aware priors, with the central principle that effective use of structural priors requires appropriate structural regularization. In our framework, the structure prior serves two key roles:
-
Anchoring dynamics: a static reference conformation provides a physically meaningful basis for modeling continuous structural variation.
-
Constraining the search space: structure-derived constraints substantially reduce the dimensionality and ambiguity of conformational exploration.
Together, these design choices allow CryoSTAR to robustly resolve heterogeneous conformations while remaining faithful to known structural information.
How CryoSTAR Works?
CryoSTAR adopts a two-phase learning framework to model conformational heterogeneity in cryo-EM datasets while explicitly incorporating structural priors.
Phase 1: Structure-regularized heterogeneity analysis
In the first phase, CryoSTAR learns a low-dimensional latent representation of continuous conformational variability using a structure-regularized variational autoencoder (VAE).
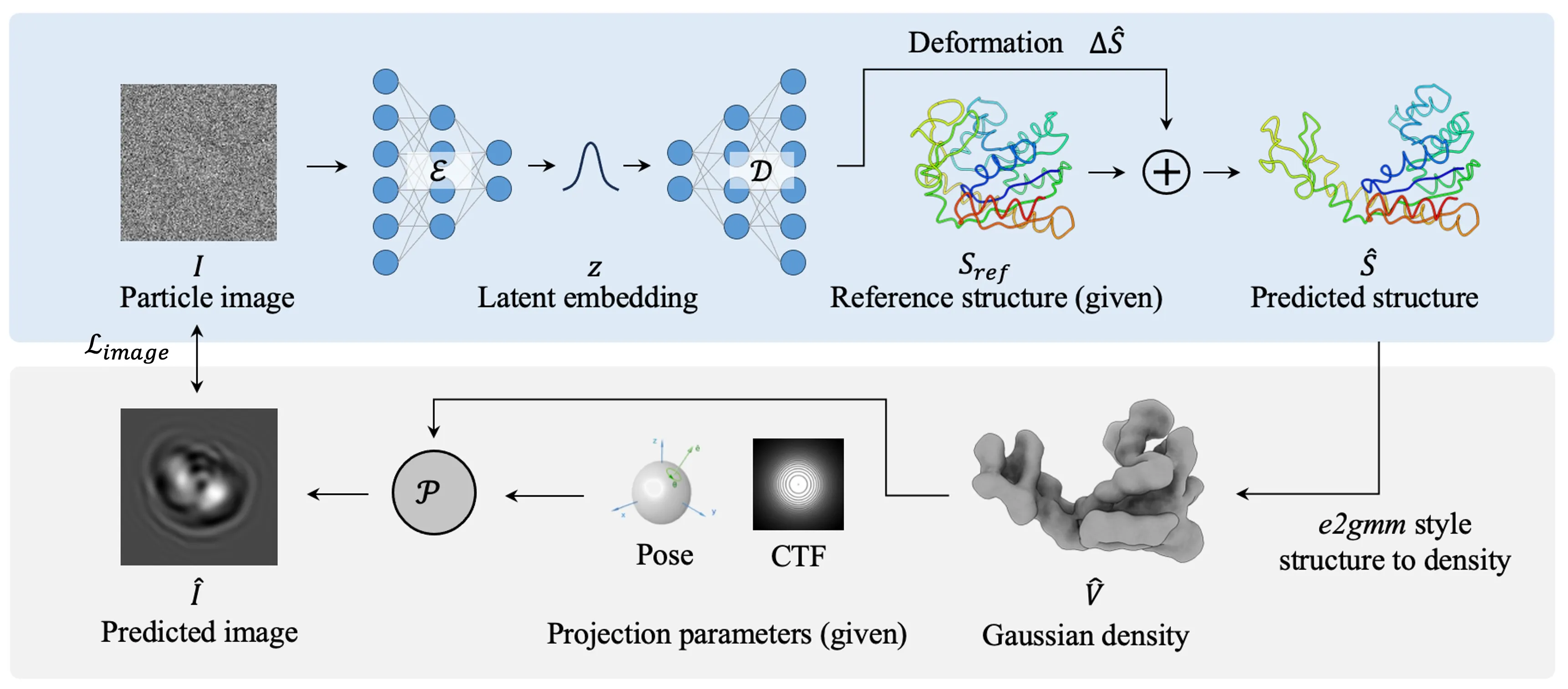
Phase 1: Structural regularized VAE.
Given an input particle image, the encoder infers a latent variable that parametrizes a deformation of a reference atomic structure. The reference structure is represented in a coarse-grained form, where each residue is modeled by a single representative atom (e.g., for proteins), resulting in an -node structure for a protein with residues.
Because cryo-EM observations are projection images of density maps rather than atomic coordinates, the predicted structure is first converted into a density representation using a Gaussian blob model, following the standard practice in e2gmm.
The density is then projected according to the known particle pose and modulated by the contrast transfer function (CTF) to generate a predicted image.
Training is performed by minimizing the reconstruction loss between the predicted and observed particle images, together with a KL divergence term that regularizes the latent posterior.
Structure-aware regularization
To ensure physically meaningful conformations and to reduce the effective search space, CryoSTAR introduces structure-aware regularization terms that explicitly constrain how the atomic model can deform:
-
Continuity constraint: Preserves the distances between adjacent residues along each chain, maintaining backbone continuity during deformation.
-
Clash avoidance: Prevents residues from approaching each other closer than an empirical covalent bond distance, avoiding unphysical bond formation.
-
Elastic network regularization: Stabilizes local secondary structure by introducing elastic constraints between residues that are spatially close in the reference structure, effectively preserving helices and β-sheets.
To mitigate reference bias introduced by elastic constraints, CryoSTAR employs an adaptive relaxation strategy. Elastic connections that consistently contribute large fitting errors are progressively relaxed or removed during training, allowing the model to accommodate alternative conformational states (e.g., open vs. closed forms) while retaining structural stability where supported by the data.
The overall objective in Phase 1 combines standard VAE losses with these structural regularization terms, yielding a latent space that captures continuous conformational variability under physically meaningful constraints.
Phase 2: Density decoding without structural bias
In the second phase, CryoSTAR focuses on producing density-based outputs for interpretation and validation.
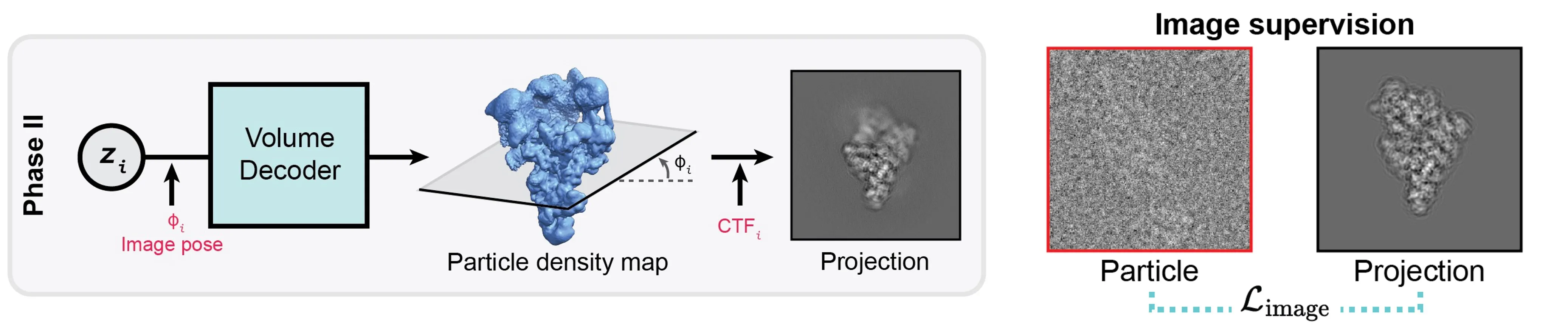
Phase 2: Density volume decoder.
Using the latent variables inferred for each particle image, a density decoder is trained to directly map latent coordinates to 3D density volumes. This decoder is trained only on particle images, without structural priors or regularization, thereby minimizing reference bias in the resulting density maps.
The generated density maps serve two purposes:
- They provide an intuitive, cryo-EM native representation of the inferred conformations.
- They enable cross-validation of the coarse-grained atomic models obtained in Phase 1.
Exploring the conformational landscape
After training, the two decoders can be jointly used to sample both coarse-grained atomic structures and corresponding density maps from any region of the latent space associated with the particle dataset. Users may explore this space using standard tools such as PCA, clustering, or other unsupervised analyses to identify dominant conformational states and their populations.
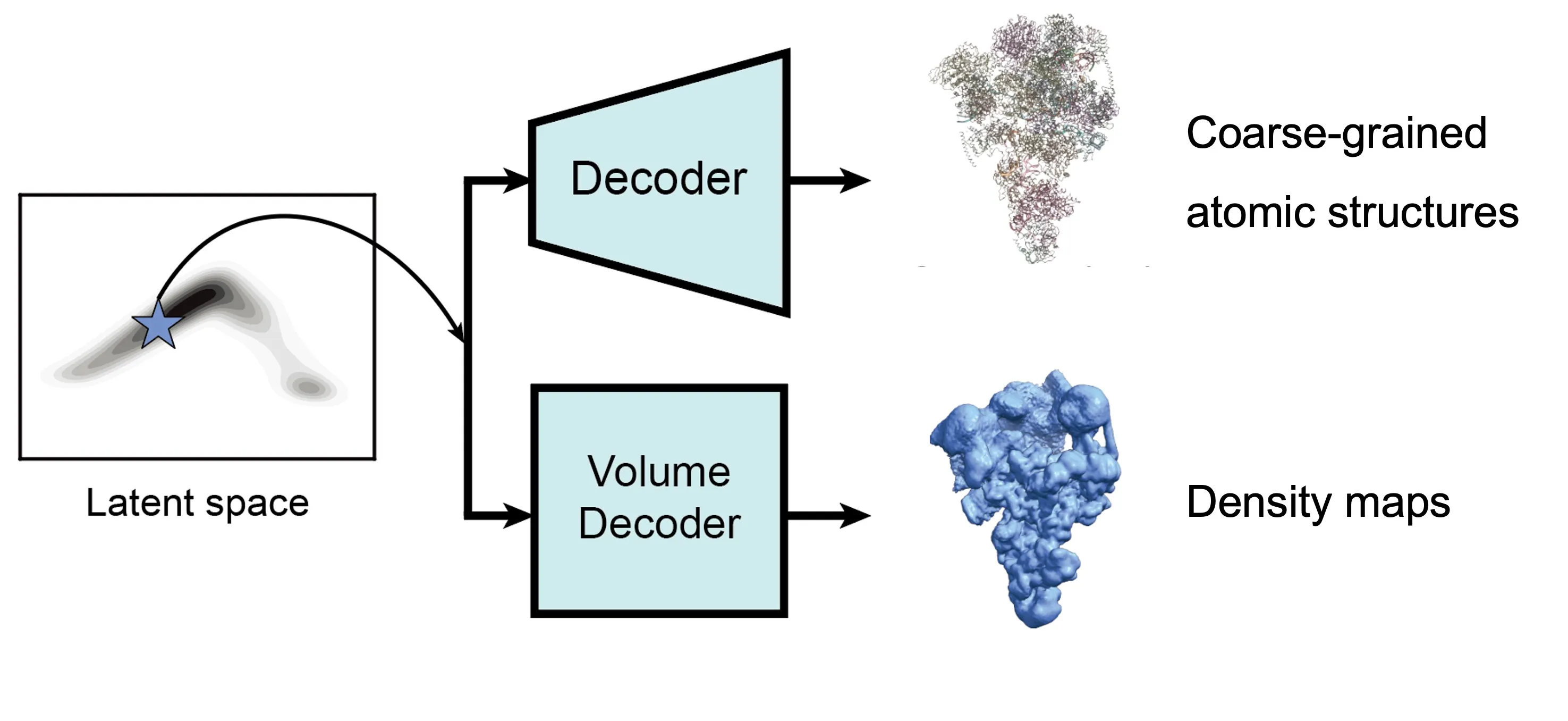
After training results.
What CryoSTAR Reveals?
The pre-catalytic spliceosome
The pre-catalytic spliceosome is a large protein-RNA complex containing more than 10,000 residues. This dataset (EMPIAR-10180) is commonly used as a benchmark for testing continuous heterogeneity algorithms.
CryoSTAR results on EMPIAR-10180.
The U4/U6.U5 tri-snRNP
The U4/U6.U5 tri-snRNP is a considerable part of the spliceosome. This dataset (EMPIAR-10073) is known to have flexible regions especially in the head and arm parts.
CryoSTAR results on EMPIAR-10073.
TRPV1 channel
TRPV1 is a 380 kDa membrane protein. In this dataset (EMPIAR-10059), particles were solubilized in nanodisc.
CryoSTAR results on EMPIAR-10059.
α-latrocrustatoxin (α-LCT)
α-latrocrustatoxin (α-LCT) is a 130 kDa spider toxin. In this dataset (EMPIAR-10827), two different conformations of α-LCT were found via discrete 3D classification, and the consensus reconstruction has a medium resolution.
CryoSTAR results on EMPIAR-10827.
Citation
If you find CryoSTAR useful, please cite:
@article{li2023cryostar,
author={Li, Yilai and Zhou, Yi and Yuan, Jing and Ye, Fei and Gu, Quanquan},
title={CryoSTAR: leveraging structural priors and constraints for cryo-EM heterogeneous reconstruction},
journal={Nature Methods},
year={2024},
month={Oct},
day={29},
issn={1548-7105},
doi={10.1038/s41592-024-02486-1},
url={https://doi.org/10.1038/s41592-024-02486-1}
}